 Biology and Evolution of Symbiosis (BESymb)
Biology and Evolution of Symbiosis (BESymb)Currently, the main lines of research we work on are focused on aphids and leeches, where we try to understand (1) the evolution of obligate nutritional endosymbionts, (2) the origin and function of gut-associated bacteria, and (3) their shared evolutionary history. Below you can find a description of funded, ongoing, and past projects.
Funded projects
Ongoing projects
The Mexican leech and its obligate nutritional symbiont as a model system for blood-feeding symbioses

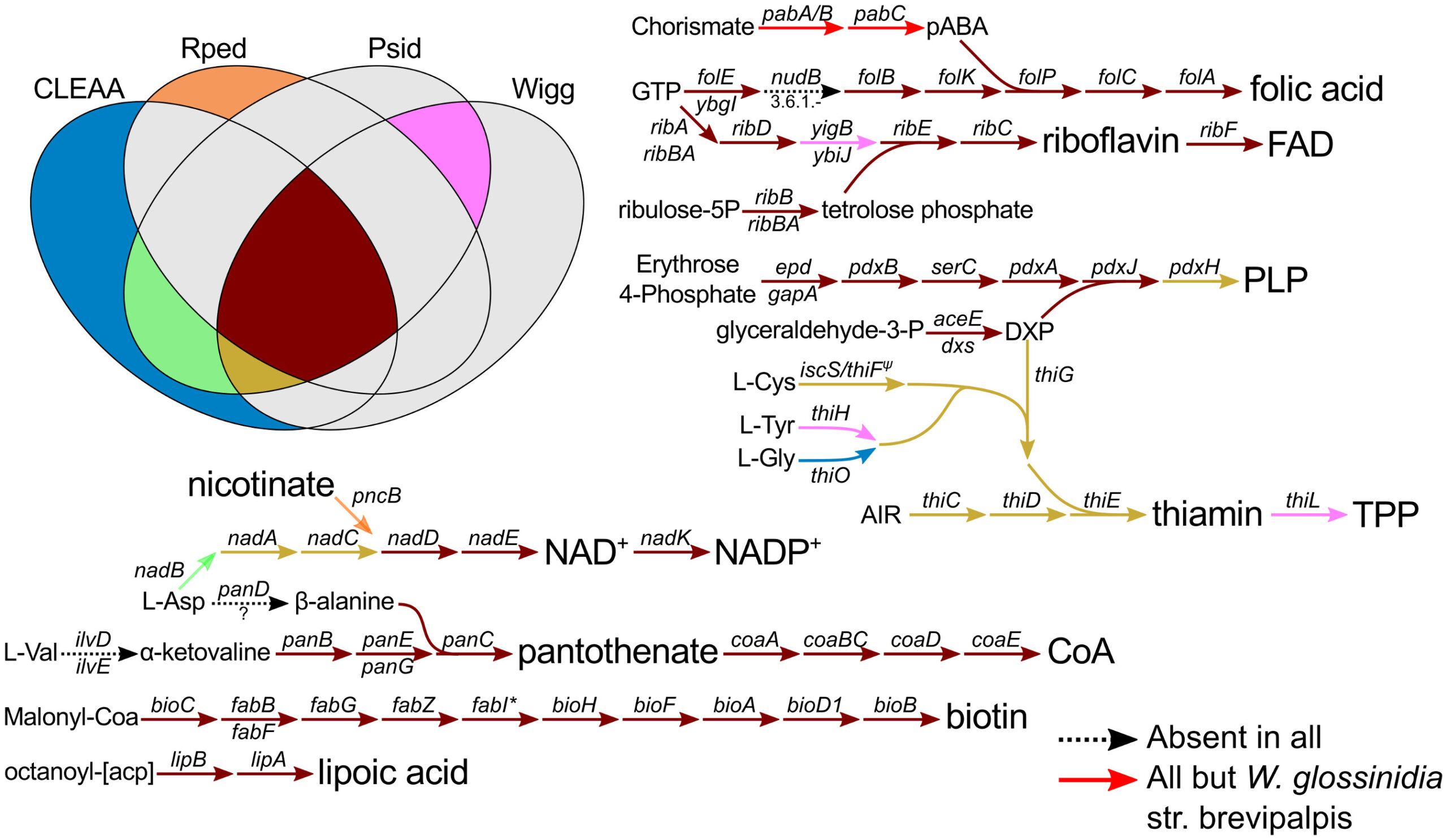
Strict blood-feeding leeches (you heard that right, there are several that do not feed on blood!) are confronted with a strong B-vitamin deficiency and thus rely on bacterial symbionts to supplement their unbalanced diet. Recent evidence showed that the Mexican leech Haementeria officinalis (A) houses, in distinct specialised organs attached to the oesophagus (B), intracellular Providencia siddallii symbionts (C, D), which have a highly reduced genome but maintain genes required for B vitamin biosynthesis (Manzano-Marín et. al. 2015). Interestingly, the metabolic steps preserved in these pathways are highly congruent with those present in obligate endosymbionts from blood-feeding arthropods, such as the Coxiella-like endosymbiont of the tick Amblyomma americanum (CLEAA), Riesia pediculicola (Rped), and Wigglesworthia (Wigg). Therefore, the mechanisms and metabolic complementation found in this system is expected to be generally applicable to nutritional symbioses in other blood-feeders, many of them important parasites, and will improve our understanding of the biology of these disease vectors.
I aim to establish the Mexican leech as a model system for studying the intricacies of nutritional blood-feeding symbioses. To achieve this, I will use different approaches:
- High quality genome sequencing of the Mexican leech Haementeria officinalis.
- Full genome sequences of the Providencia symbionts of several Haementeria species.
- Genome-based metabolic inference of the host-symbiont “cross-talk” for the production of B-vitamins in the Providencia–Haementeria symbiosis.
This project was financed by a Marie Skłodowska-Curie Postdoctoral Fellowship (H2020-MSCA-IF-2018 LEECHSYMBIO, grant agreement 840270), and (despite COVID-related setbacks and re-worked research aims) was being developed at the Centre for Microbiology and Environmental Systems Science (CMESS) from the University of Vienna with the collaboration of Dr. Alejandro Oceguera Figueroa from the Biology Institute (IB) at UNAM, Mexico.

Genomics and evolution of leeches (Annelida: Hirudinida)
Leeches are a group of annelids of more than 700 currently described species with a widespread blood-feeding habit. They are found in a variety of environments (freshwater, estuaries, marine ecosystems, and even land). While haematophagy (or blood-feeding) is what makes this group most (in)famous, not all members feed on blood. Some of these non-blood feeders are predatory, while others suck the fluids out of invertebrates. Until recently, very little was known about leech genomics, with only the genome of the non-blood-feeding glossiphoniid Helobdella robusta. Through different collaborations with leech taxonomists, we are aiming at expanding our knowledge on these fascinating and diverse critters, including their genetic diversity, anticoagulant repertoires, and genomic-traits that have contributed to their intriguing biological features.
– Medicinal leeches

Haematophagous medicinal leeches have historically been used as medical tools of antiquity most notably in the practice of bloodletting as a “cure” for a number of afflictions. In modern science-based medicine, they are used as agents to stimulate circulation of blood in skin grafts, finger reattachment, and other tissue threatened y postoperative venous congestion. The notable ability of blood-feeding is facilitated by anticoagulant enzymes present in the leech’s saliva, which avoids blood flow from being interrupted while the leech feeds. These anticoagulant proteins are one of the evolutionary innovations, along with morphological ones such as specialised mouth parts, that have repeatedly evolved to facilitate blood-feeding across haematophagous animals (Iwama et. al. 2021).

Along with Dr. Sebastian Kvist and team (Swedish Museum of Natural History) as well as Prof. Peter Trontelj (University of Ljubljana), we sequenced, assembled and analised the genome of the “epic” European medicinal leech Hirudo medicinalis (Kvist et. al. 2020). We aimed at determining the repertoire of anticoagulant proteins from this historical and medically important leech. We managed to not only identify 18 well-known anticoagulant enzymes and their previously unknown copy number in the leech’s genome but also a previously unknown set of 23 putative anticoagulants. In all, the large anticoagulant repertoire that H. medicinalis encodes suggests that the potency of the leech’s saliva in counteracting the clotting cascade of blood is dependent on a larger than previously known cocktail of proteins that have several different targets in the cascade.
We are currently aiming for the sequencing of further and diverse leech species with different goals for each set of genomes. Stay tuned for updates!
Origin and Evolution of co-obligate symbioses across aphids (Hemiptera: Aphididae)
Typically, aphids house the obligate nutritional bacterial symbiont Buchnera inside specialised cells called bacteriocytes. Buchnera supplies the aphid with essential amino acids and vitamins thus insuring the correct development of its host. However, some Buchnera lineages have lost the ability to fulfill this role, either triggered or rescued by new and younger endosymbionts. One such case are the aphid species from the Lachninae subfamily, where an ancient loss of the riboflavin biosynthetic genes in the genome of Buchnera was accompanied by the acquisition of a co-obligate partner, putatively Serratia symbiotica (Manzano-Marín et. al. 2017). However, co-obligate symbioses are not restricted to this subfamily, and examples of these have been previously suggested mainly by microscopic studies.

Through whole genome sequencing, we have reconstructed the genomes of the co-obligate endosymbionts from several aphid species belonging to different subfamilies (Manzano-Marín et. al. 2023). We have corroborated that these co-obligate symbionts indeed supplement the metabolic deficiencies found in Buchnera, mainly that of biotin and riboflavin. Not surprisingly, they have evolved genomes with similar core metabolic capabilities, with some even bringing new ones to the symbiotic consortium. These co-obligate symbionts have evolved from diverse facultative symbiotic taxa associated to aphids as well as free-living bacterial strains. These findings show that co-obligate symbiosis in aphids is more widespread than previously thought. This suggests a fragile mono-symbiotic association between the aphid host and its Buchnera symbiont, whose highly degenerated genome could undergo simple metabolic losses which could lead to a “lucky” secondary symbiont establishing as a co-obligate one.
This project was financed by a Marie Curie Postdoctoral Fellowship (FP7-PEOPLE-2013-COFUND AgreenSkills+, Grant agreement 609398) during my first postdoctoral stay at the Centre de Biologie pour la Gestion des Populations (CBGP) under the supervision of Dr. Emmanuelle Jousselin. The project is currently continuing in collaboration with Dr. Jousselin.

Evolution of the gammaproteobacterial symbiont Serratia symbiotica in Cinara aphids
Aphid species from the Lachninae subfamily have been found to harbour Buchnera strains that have ancestrally lost the capacity to synthesize biotin and riboflavin (Manzano-Marín et. al 2016), two essential B vitamins for the aphid host. For the provision of these nutrients, Lachninae aphids and their Buchnera now rely on different co-obligate endosymbionts, most often S. symbiotica (Manzano-Marín et. al. 2017). Accordingly, Cinara species (Aphididae: Lachninae) have been consistently found to host an additional bacterial co- obligate symbiont, most commonly S. symbiotica (Meseguer et al. 2017). An ancestral reconstruction of the symbiotic associations of Cinara with fixed additional symbionts suggests that S. symbiotica was likely the original co-obligate endosymbiont, but has been replaced several times by other bacterial taxa. These S. symbiotica symbionts have radically different genomes, with sizes ranging from 1.2 to 2.4 Mbp and G+C contents from 32 to 52%. Additionally, they have found to have different tissue tropisms: resembling the localisation, distribution, and cellular shape of both facultative and obligate symbionts, depending on the host species (Manzano-Marín et. al. 2017).
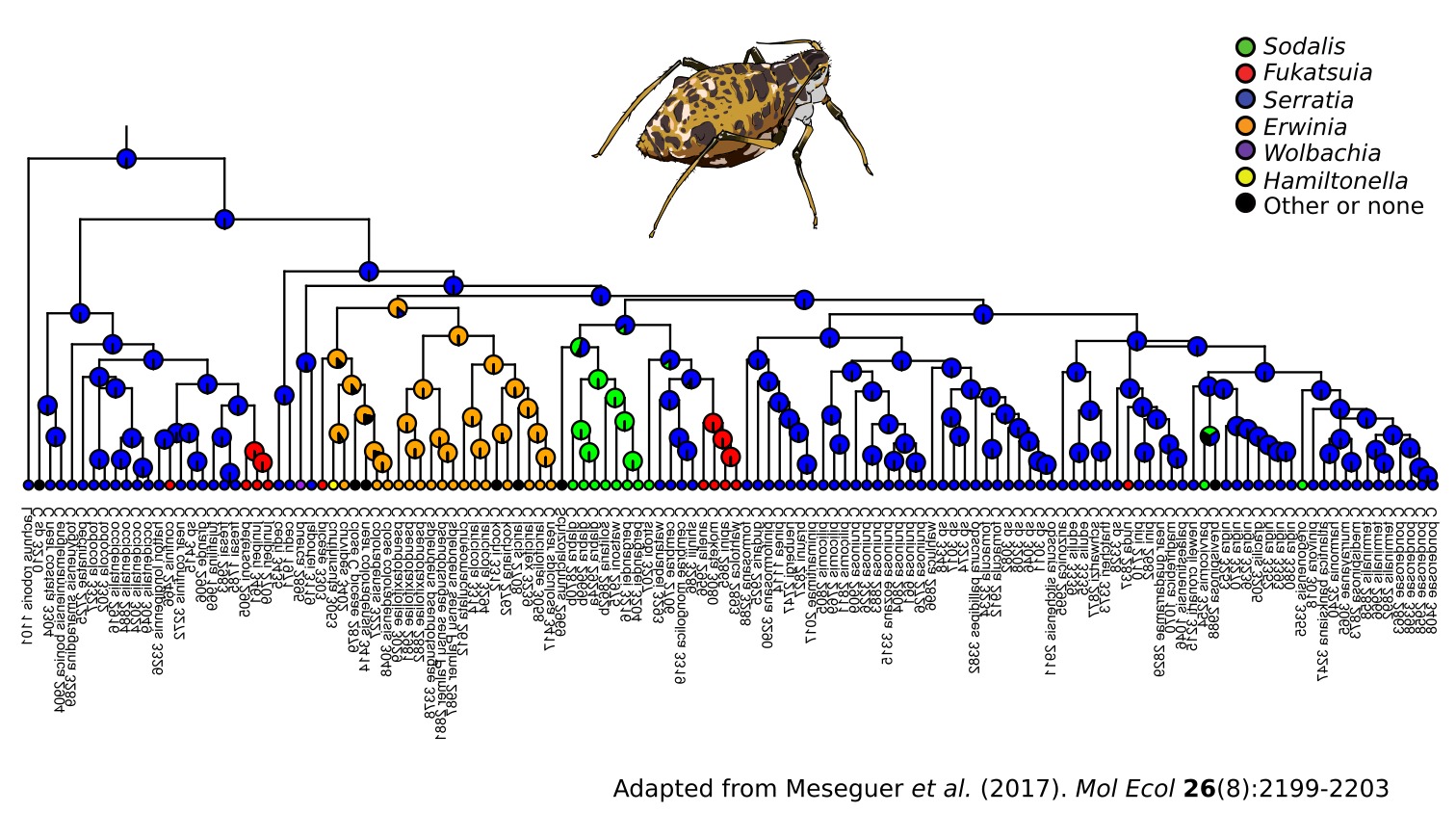
Through whole genome sequencing of symbiont-enriched DNA samples of over 60 Cinara aphids, we have reconstructed the genomes of Buchnera–S. symbiotica endosymbiont pairs. Through phylogenetic, cospeciation, and comparative genomic analyses, we aim to understand the evolutionary history of this widespread secondary co-obligate symbiont as well as the alternative routes each co-obligate lineage has followed to establish a beneficial cooperative association with Buchnera in their aphid hosts.
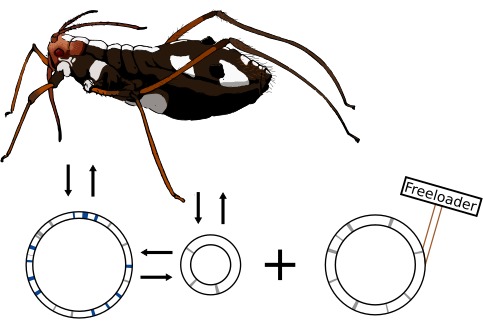
Intermediate results from this project have already uncovered interesting mechanisms of symbiont replacement and the emergence of new symbiotic relationships. In the aphid Cinara strobi, we found that the previously co-obligate S. symbiotica has putatively already been metabolically replaced by a “younger” Sodalis symbiont (Manzano-Marín et. al. 2018). Nonetheless, the S. symbiotica symbiont persists, being present in 100% of the samples analysed, but lacks intact routes for all essential amino acids and B vitamins. Strikingly, it has evolved a rather “deserted” genome, with a staggering low coding density of 26.3% (635 CDSs), despite having a moderately reduced genome of 2.41 Mbp. Our results suggest that S. symbiotica has become a “freeloader” which likely evolved from an ancient co-obligate lineage.
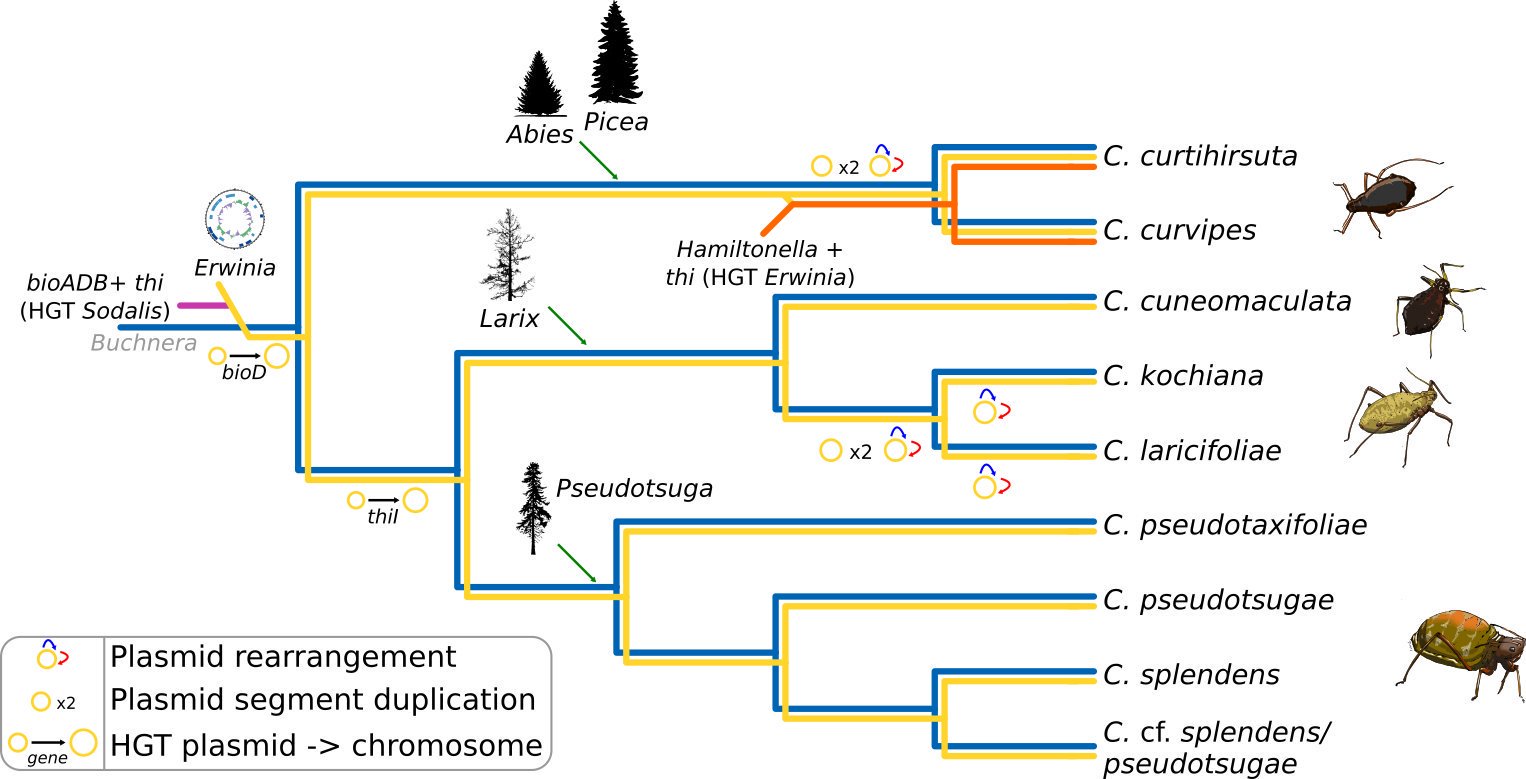
In a particular group of Cinara, S. symbiotica has been ancestrally replaced by Erwinia bacteria. Through whole genome sequencing, fluorescence in situ hybridization (FISH), and comparative genomics, we found that the new Erwinia symbiont (Candidatus Erwinia haradaeae), is located to the bacteriome in close proximity to Buchnera, has evolved a small A+T-rich genome, and it is capable of complementing Buchnera‘s metabolic deficiencies (Manzano-Marín et. al. 2020). Moreover, this new symbiont bring a new nutritional role to the symbiotic consortium: the biosynthesis of the essential B vitamin thiamin. Most of genes involved in this metabolic pathway (except for thiL) have been horizontally transferred from a Sodalis-related bacterium. This same horizontal transfer pattern was found for the biotin biosynthetic genes bioA, bioD, and bioB; which convert KAPA to biotin. Moreover, these genes have been further transferred to a third co-obligate symbiotic Hamiltonella partner in the common ancestor of C. curtihirsuta + C. curvipes. Indeed, our genomic enquiry suggests that thiamin biosynthesis genes might be pivotal in the establishment of both E. haradaeae and then Hamiltonella, raising questions about the importance of this nutrient for the insect host.
This project was financed by a Marie Curie Postdoctoral Fellowship (FP7-PEOPLE-2013-COFUND AgreenSkills+, Grant agreement 609398) during my first postdoctoral stay at the Centre de Biologie pour la Gestion des Populations (CBGP) under the supervision of Dr. Emmanuelle Jousselin. The project is currently continuing in collaboration with Dr. Jousselin.
Past projects (Coming soon…)
Evolution and comparative genomics of di-symbiotic systems in aphids from the Lachninae subfamily
Coming soon…

Genome reduction in the gammaproteobacterial endosymbiont Serratia symbiotica
Coming soon…